
Mohamed Hamdy Amar
National Key Laboratory of Crop Genetic Improvement, Huazhong Agricultural University, Wuhan 430070, China
LiveDNA: 20.5385







ABSTRACT
In the present study, we sought to determine whether one simple criterion, sequence divergence, can reasonably guide in phylogenetic across a broad scale in Citrus germplasm. Comparative investigation on the performance of the SSR and SRAP markers was conducted in phylogenetic analysis across sequence analysis of the PCR product in the genus Citrus and its relatives. The maximum composite likelihood model was used for pairwise distance calculation. To determine whether there is a difference depending on the method of choice. Somehow, phylogenetic trees were constructed using two algorithms Neighbor Joining (NJ) and Maximum Parsimony (MP) via MEGA 4 software. In contrast some differences in the positioning of some genotypes were observed in the phylogenetic trees created using the two models and the dendrogram from MP across SRAP sequence was the most congruent with Swingle and Reece’s treatment of the subfamily Aurantioideae. The results of the present study suggest that evaluation of SRAP variation at the sequence level can be effective than SSR variation in exploring the evolutionary relationships among Citrus species. These results were the new information for future study on Citrus breeding programs such as germplasm characterization, screening of zygotic and nuclear seedlings and developing sequence divergence in Citrus and its relatives.
PDF Abstract XML References Citation
Received: November 05, 2011;
Accepted: December 28, 2011;
Published: February 15, 2012
How to cite this article
Mohamed Hamdy Amar, 2012. Comparative Analysis of SSR and SRAP Sequence Divergence in Citrus Germplasm. Biotechnology, 11: 20-28.
DOI: 10.3923/biotech.2012.20.28
URL: https://scialert.net/abstract/?doi=biotech.2012.20.28
DOI: 10.3923/biotech.2012.20.28
URL: https://scialert.net/abstract/?doi=biotech.2012.20.28
INTRODUCTION
Citrus is one of the world’s important fruit crops which is widely grown in most areas with suitable climates between latitude 35°N~35°S (Liu and Deng, 2007. The total global production reported to be 7.4 million metric tons in 2009-2010 (http://faostat.fao.org/site/339/default.aspx). Citrus and the closely related genera are partially sexually compatible in varying degrees; they are primarily diploid with a few known triploids and occasional tetraploid forms (2n = 2x = 18) and they possess fairly small genomes (e.g., sweet orange has been said to be around 367 Mb, or approximately three times that of Arabidopsis). As such, the citrus species should be amenable to many of the commonly used techniques and approaches related to genomic research, including genetic and physical mapping, full genome sequencing and functional genomics studies aimed at unravelling the complexities of key traits of interest (Talon and Gmitter, 2008).
With the rapid development of molecular biology studies of Citrus germplasm identification and genetic diversity offer, numerous reliable molecular marker information by means of AFLPs, SSRs, ISSRs, IRAPs and SRAP (Chen et al., 2006; Uzun et al., 2009).
In recent years, a tally of expressed sequence tags (ESTs) available from the NCBI EST database is refreshed weekly and displayed on the International Citrus Genomics Consortium website (http://int-Citrusgenomics.org/). Globally, EST-derived microsatellites have been observed to have high conserved flanking sequences among related species. Meanwhile the increase in available DNA sequence information, particularly ESTs has provided new opportunities for development of molecular markers for Citrus sp. (Palmieri et al., 2007). EST sequences have been produced by several research groups from a number of Citrus species (Terol et al., 2007). Hence, EST-SSR markers are powerful tools to investigate genetic diversity and genome mapping in Citrus (Luro et al., 2008).
On behalf of Sequence Related Amplified Polymorphism (SRAP), it is a PCR-based marker system as described by which aimed for the amplification of Open Reading Frames (ORFs) (Li and Quiros, 2001). The SRAPs is a simple and efficient marker system that can be adapted for a variety of purposes in different crops, including germplasm identification, map construction, gene tagging, genomic and cDNA fingerprinting and map based cloning. It has several advantages over other systems. It discloses numerous co-dominant markers; targets Open Reading Frames (ORFs) and allows easy isolation of bands for sequencing (Uzun et al., 2009). SRAPs were easily amplified in crops such as potato, rice, lettuce, cotton, citrus and some other genera in subfamily Aurantioideae (Dong et al., 2010).
Advances in DNA sequencing techniques have allowed the extensive use of DNA fragments, especially the analysis of DNA sequence variation which is the major importance in genetic studies like genome mapping, gene tagging and pedigree analysis (Kota et al., 2001). Sequence divergence is the direct result of nucleotide substitution, which occurs according to the properties of specific genes (invariable sites and transition/ transversion ratio due to selection) and genomic environment (nucleotide and amino acid bias) (Makowsky et al., 2010).
Consequently, documenting changes at the sequence level enables more precise analysis of intra- and interspecific polymorphism and also offers a view of how any given repeat structure has evolved. So, it is very handy to use SSR and SRAP from genomic clones to detect genetic differences and to undertake phylogenetic reconstructions among diverse accessions. (Guo et al., 2005).
The aims of the present study were to characterize the nature of sequence variation in SSR and SRAP to analyze how any given repeat structure has evolved. Furthermore to conducted comparative investigations on the performance of the two molecular markers in phylogenetic analysis across sequence analysis of the PCR product in the genus Citrus and its relatives. Finally, to understand the evolutionary history of 24 Citrus species and their putative ancestors based on sequence information of SSR and SRAP markers.
MATERIALS AND METHODS
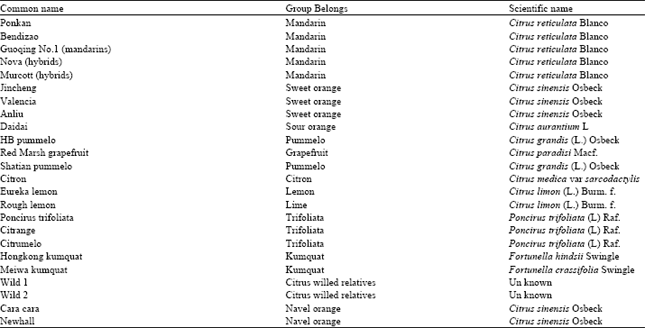
Plant materials: Twenty four genotypes of Citrus species were collected from the National Center of Citrus Breeding (NCCB), Huazhong Agricultural University (HZAU), Wuhan, China; the materials were originally collected from different parts of China and other countries. These genotypes belonging to the major groups of Citrus and its relatives (mandarin, sweet orange, sour orange, pummelos, citron, lemon, trifoliate, kumquats, wild relatives and navel orange) which summarized in Table 1.
Methods
DNA isolation: Total genomic DNA was isolated from fresh leaves of 24 different Citrus varieties following the procedure previously described by Cheng et al. (2003) The quality and concentration of the DNA samples were checked in a UV-1601 spectrophotometer (Shimadzu, Japan) and sample concentrations were adjusted to 50 n μL-1.
SSR analysis: SSR primers were designed using EST analysis according to Amar et al. (2011). Ten primer pairs of SSR were designed according to the sequences flanking of SSR repeats, thus, to chosen the best combinations primers were selection for sequencing. The primers were synthesized by Sangon (sangon. Com, China).
| Table 1: | List of 24 Citrus and its relatives used in the present study |
 | |
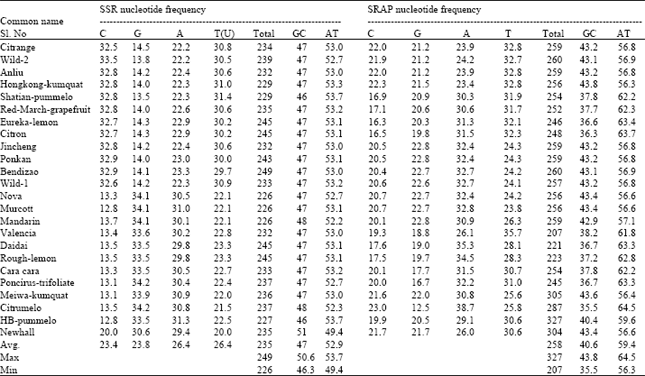
| Table 2: | Nucleotide Frequencies for SSR and SRAP sequence |
 | |
Primer annealing temperatures varied from 53 to 61°C depending on the base composition of the primers. In general, the difference in annealing temperatures between the forward and the reverse primer did not exceed 3°C. The lengths of amplified products were between 150 and 350 base pairs (bp). SSR amplification was conducted in 20 μL of reaction mixture containing 10x PCR buffer, 2.0 mmol MgCl2, 0.15 mmol dNTP, 4 pmol of each primer (forward and reverse), 50 ng templates DNA and 1.0 U Taq DNA polymerase. The amplification reaction procedure was as follows: After denaturation at 94°C for 4 min, the reaction mixture was subjected to amplification for 10 cycles consisting of 30 sec at 94°C, 30 sec at 66°C and 45 sec at 72°C, followed by 30 cycles consisting of 30 sec at 94°C, 30 sec at 55°C and 45 sec at 72°C with a final extension at 72°C for 10 min. The amplification products were separated by electrophoresis in a 6% polyacrylamide gel visualized by a simplified silver staining method (Xu et al., 2002).
SRAP analysis: SRAP primer combinations were screened using 36 different combinations which employed using six forward and reverse primers (Table 2). Across whole primers screened only 10 SRAP combinations primers gave highly levels of polymorphism. Thus, to chosen the best combinations primers were selection for sequencing. For PCR amplification, each 20 μL PCR reaction mixture consisted of 30 ng genomic DNA, 2.0 μL of 10xPCR buffer, 2 mM of MgCl2, 200 mmol dNTPs, 0.2 mmol for each primer and 1 U Taq polymerase. PCR cycling parameters included 4 min of denaturing at 94°C, five cycles of three steps: 1 min of denaturing at 94°C, 1 min of annealing at 35°C and 1 min of elongation at 72°C. In the following 35 cycles the annealing temperature was increased to 50°C and for extension, one cycle 7 min 72°C. A 100 bp DNA ladder was used as molecular standard in order to confirm the appropriate SRAP markers. The amplified SRAP fragments were separated and visualized with the same procedure as for SSR.
Cloning of PCR products: Two primers were selected for sequencing data for SSR and SRAP. Primer (MHA 48) was chosen for SSR markers with the sequence (Forward 5' CAACCGTTCCTGACTCCATT 3' and Reveres 5' AAGTGTTTTCGAGGTGGGTG 3') and primer Me10/Em01 was selected for SRAP markers with the sequence Forward 5’TGAGTCCAAACCGGAAA3’ and Reveres 5’GACTGCGTACGAATTAAT) which gave the highest polymorphism among citrus genotype under this study.
The PCR products were subjected to electrophoresis on a 1.5% agarose gel, stained with ethidium bromide and visualized under ultraviolet (UV) light. The PCR fragments of each sample were excised and purified from the gel using E.Z.N.A® Gel Extraction Kit (Omega, USA). For cloning, the PCR fragments were ligated to pMD18-T easy vector using the appropriate kit (TaKaRa, Japan) and the ligation product was transformed into Escherichia coli DH-5α-competent cells. The recombinant clones were selected on Liquid Broth (LB) plates containing ampicillin. Cloned PCR products were sequenced by the Uni-Gene Company (Shanghai, China).
Sequencing analysis: Vector sequences were removed and the sequence was aligned using the computer program Clustal X version 1.81 (Thompson et al., 1997) with manual adjustments as necessary. Gaps were positioned to minimize nucleotide mismatches. The MEGA program version 4.0 (Tamura et al., 2007) was employed to estimate GC content, nucleotide substitution, nucleotide diversity (π) and cluster analysis among the 24 citrus genotypes.
Phylogenetic analysis: Phylogenetic and molecular evolutionary analyses were conducted using MEGA 4 software version 4. Maximum composite likelihood model was used for pairwise distance calculation. Phylogenetic trees were constructed using two algorithms: Neighbor Joining (NJ) and Maximum Parsimony (MP) (Apostolidis et al., 2001).
RESULTS
Sequencing analysis for SSR and SRAP: In the present study, two primers from SSR and SRAP were selected with high performance for sequencing among the twenty four genotypes of Citrus species (Table 1). A total of 48 PCR products were generated and the length of the PCR product for SSR and SRAP markers were ranged from (150 to 350 bp) and for (600 to 700 bp), respectively. While the total numbers of nucleotide sequences were 5650 and 6210 for SSR and SRAP, respectively.
Nucleotide composition was obtained and the results for the nucleotide frequencies of SSR and SRAP are summarized in Table 2. The highest numbers of nucleotide for SSR sequence was observed in Bendizao (249 bases) whereas, the lowest one were recorded in Nova, Murcott and Mandarin (226 base). Furthermore, the averages of GC (47.1%) and AT (52.9%) content were calculated using MEGA 4 program. The maximum nucleotide frequency for GC content (51%) was observed in Newhall, while HB-pummelo was the maximum nucleotide frequencies for AT content (53.7%). In contrast, the lowest GC content (46%) and AT content (49.4%) were observed in Shatian-pummelo and Newhall, respectively.
On behalf of the nucleotide base composition of SRAP sequence, Citron was the maximum numbers of nucleotide (327 bases) whereas, Eureka-lemon was the minimum numbers of nucleotide (207 bases). However, the averages of GC (40.6%) and AT (59.4%) content were conducted. The highest percentages of GC (43.8%) and AT (64.5%) content were recorded in Wild-1 and Bendizao, respectively. On the other hand, the lowest GC content (35.5%) and AT content (56.3%) were observed in Bendizao and Wild-1.
The Tajima's Neutrality test (Tajima, 1989) was performed (Table 3) to calculate the nucleotide diversity value (π) using MEGA4 program. All positions containing gaps and missing data were eliminated from the dataset. Highest Tajima test statistic (D = 4.55) and nucleotide diversity value (π = 0.43) were observed in SRAP sequence when compared with SSR sequence (D = 3.14) (π = 0.28) (Table 3).
Phylogenetic analysis: Based on the sequence data of the flanking regions of SSR and SRAP sequence data, a phylogenetic tree were constructed using the two algorithms, NJ and MP methods to determine whether there is a difference depending on the method of choice.
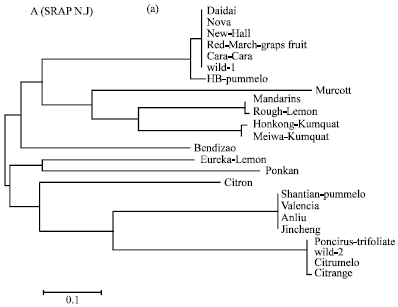
The evolutionary distances for SSR sequence were computed using the Maximum Composite Likelihood method (Tamura et al., 2004). The phylogenetic analysis of SSR sequence data among 24 Citrus species is shown in Fig. 1a and b. NJ distance phylogram clustered Citrange, Wild1, Hongkong-kumquat, Eureka-lemon, Citron, Red-March-grapefruit, Anliu, Jincheng, Bendizao and Ponkan into a distinct clade (clade 1), while Shatian-pummelo, Wild-2 and Newhall both fell into a separate clade (clade 2, clade 3 and clade 4). However, Poncirus-trifoliata and Citrumelo were grouped in a separate clade (clade 5). For the clade (6), HB-pummelo and Meiwa-kumquat were compress together. In contrast, Mandarin, Valencia, Daidai, Rough-lemon, Cara Cara, Nova and Murcott were sheared together in a separate clade (clade 7).
On behalf of the Maximum Parsimony (MP) analysis for SSR sequence, Ponkan, Eureka-lemon, Jincheng, Citron, Jincheng, Red-March-grapefruit, Citrange, Anliu, Hongkong-kumquat, Bendizao, Wild1 and Shatian-pummelo were assembled together in the first main cluster while Wild-2 and Newhall were separated individually. In the second main cluster, Poncirus-trifoliata, Citrumelo and Meiwa-kumquat were grouped together in a separate clade (clade 1). Moreover, Nova and Murcott were sheared together in the second clade. Likewise, Valencia, Mandarin, Rough-lemon, Cara Cara, Daidai and HB pummelo were comprised together in clade 3.
With respect to SRAP sequence, evolutionary relationships were evaluated by means of the (NJ) and (MP) methods and summarized in Fig. 2 a and b.
| Table 3: | The evolutionary analyses using Tajima test statistic among SSR and SRAP sequence |
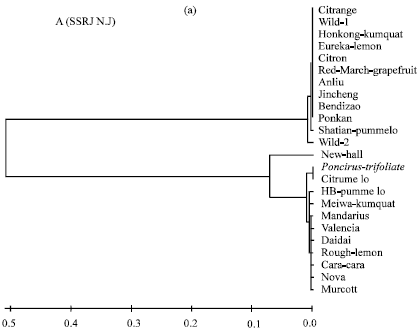 | |
| Fig. 1a: | Evolutionary relationships of 24 citrus species as inferred for the Neighbor-Joining method (N.J) tree model using SSR sequence. |
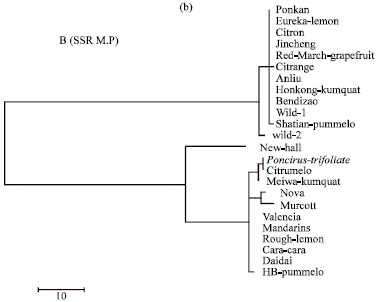 | |
| Fig. 1b: | Evolutionary relationships of 24 citrus species as inferred for the Maximum Parsimony (M.P) tree model using SSR sequence |
Within the Neighbor Joining (NJ) distance clustered Daidai, Nova, Newhall, Cara Cara, Red-March-grapefruit and Wild1 were cluster together in the first clade. While the two pairs Mandarin and Rough-lemon and Hongkong-kumquat and Meiwa-kumquat were shared in a separate clade (clade 2). Within the second cluster, Eureka-lemon and Ponkan were formed together in a sister clade (clade 3). Moreover, Shatian-pummelo, Valencia, Anliu and Jincheng were compressed in a separate clade (clade 4). Likewise, Poncirus-trifoliate, Wild-2, Citrange and Citrumelo were sheared in the last clade. On the other hand, Murcott, Bendizao, HB pummelo and Citron were excluded individually.
 | |
| Fig. 2a: | Evolutionary relationships of 24 citrus species as inferred for the Neighbor-Joining method (N.J) tree model using SRAP sequence |
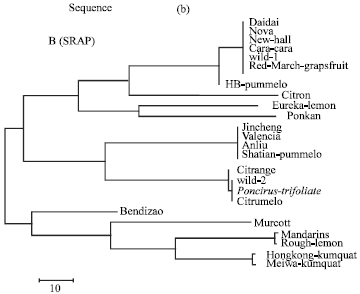 | |
| Fig. 2b: | Evolutionary relationships of 24 citrus species as inferred for the Maximum Parsimony (M.P) tree model using SRAP sequence |
For the Maximum Parsimony (MP) analysis of SRAP sequence, Daidai, Nova, Wild 1, Cara Cara, New Hall, and Red-March-grapefruit were grouped jointly in the first clade. However, Eureka-lemon and Ponkan were sheared together in the second clade. Furthermore, Jincheng, Valencia, Anliu and Shatian-pummelo were compressed in the third clade. In addition, Poncirus-trifoliate, Wild 2, Citrange and Citrumelo were grouped together in the forth clade. Within the second cluster, the two pairs Mandarin and Rough-lemon and Hongkong-kumquat and Meiwa-kumquat were sheared in a sister clade (clade 5 and 6). Nevertheless, the hybrid species HB pummelo, Citron, Bendizao and Murcott were excluded individually.
In contrast some differences in the positioning of some genotypes were observed in the phylogenetic trees model and Maximum Parsimony model for SRAP sequence was the most congruent with Swingle and Reece’s treatment of the subfamily Aurantioideae than the other models were tested through SSR and SRAP sequences in Citrus.
| Table 4: | Maximum composite likelihood estimate of the pattern of nucleotide substitution of substitution matrix for SSR and SRAP sequence |
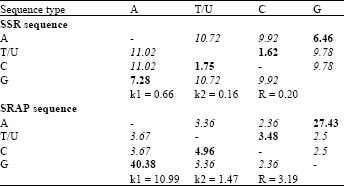 | |
| Rates of different transitional substitutions are shown in bold and those of transversional substitutions are shown in italics | |
DISCUSSION
Citrus taxonomy and phylogeny is often the subject of controversy because of the high diversity of phenotypic characters, their long history of cultivation and complex reproduction system. Indeed, a self- incompatibility gene, facultative apomixes, sterility gene, and wide sexual compatibility between Citrus sp. and related genera can all be found (Froelicher et al., 2011). In the present study, we sought to determine whether one simple criterion, sequence divergence, can reasonably guide gene choice in phylogenetic across a broad scale. Using both natural and simulated datasets, our results show that certain levels of sequence variation are similar to the relations of the previous evolution (Kyndt et al., 2010; Froelicher et al., 2011). In this article, one of our goals was to elucidate the phylogenetic placements of 24 Citrus species and its relatives using the sequence divergence across SSR and SRAP markers, in addition to explain the superior tree model for sequence identification via Mega 4 program.
In Table 4 the substation pattern and rates were estimated to compare the similarity matrix under the Tamura-Nei 93 test model (Tamura and Nei, 1993). The each entry is the probability of substitution (r) from one base (row) to another base (column). Rates of different transitional substitutions are shown in bold and those of transversionsal substitutions are shown in italics. The transition/transversion rate ratios and the overall transition/transversion bias for SSR sequence were k1 = 0.661 (purines), k2 = 0.163 (pyrimidines) and R = 0.20, respectively. Meanwhile the transition/transversion rate ratios and the overall transition/transversion bias for SRAP sequence were k1 = 10.99 (purines), K2 = 1.47 (pyrimidines) and R = 3.19, respectively. The results indicated that transitional substitution was highest value in SRAP compared to SSR sequence. This reflects that transitions are more dominant than transversion in Citrus germplasm across SRAP markers.
| Table 5: | The Maximum Likelihood fits using Tamura-Nei 93 model across 24 different nucleotide of SSR and SRAP sequence |
Consequently, SRAP sequence divergence to be used as a significant molecular marker for classification and identification at the species level and beyond. In count of the nucleotide frequencies, (A 30%) and (T 26%) were highly frequency in SRAP sequence compare with SSR sequence. in contrast, the frequency of C and G were convergent among SSR and SRAP sequence (Table 5).
The phylogenetic trees were constructed using two algorithms (NJ) and (MP) to determine whether there is a difference depending on the method of choice. On the contrary, some differences in the positioning of some genotypes were observed in the phylogenetic trees created using different models and the dendrogram from MP was the most congruent with the previous studies of Citrus (Uzun et al., 2009; Biswas et al., 2010; Amar et al. 2011). In the current revise, grapefruit (C. paradisi) has been proposed to be of hybrid origin, with pummelo as mother and sweet orange as father (Gmitter, 1995; Moore, 2001) and subsequent backcrossing with pummelo (Pang et al., 2007; Dianxiang and Mabberley, 2008; Mabberley, 2008). Sweet orange (C. sinensis) is thought to be a natural hybrid between predominantly C. reticulate and some C. maxima (Scora, 1975; Barrett and Rhodes, 1976). Molecular data already confirmed that the genome of sweet orange is derived from pummelo (Nicolosi et al., 2000; Barkley et al., 2006). While the hybrid genotypes daidai (C. aurantium sour orange), nova (C. reticulate x Cparadisi) and navel orange (C. sinensis) are closely related with pummelo. Mandarin played an important role in the evolution of cultivated Citrus. In addition, phylogenetic analysis by Scora (1975), Barrett and Rhodes (1976) suggested that mandarin was one ancestral parent of both sweet orange and sour orange. Nuclear marker analysis supported this hypothesis and showed that the mandarin gene pool also contributed to lemon (Froelicher et al., 2011). The citron genotype contained only (C. medica.), this species did not transmit its cytoplasm to other species but played an important role as a male parent. Indeed, using nuclear and cytoplasmic markers (Nicolosi et al., 2000). Our results confirm that pummelo was the maternal parent of C. sinensis and played a part in the parentage of many of the cultivars of Citrus, this in line with the results of Yamamoto et al. (1993), Nicolosi et al. (2000) and Barkley et al. (2006). Additionally, our results confirm that C. sinensis and trifoliate shared individually in a separated clade forming a sister relationship. Recently, similar results were also documented by Froelicher et al. (2011) who support that usage of sequence divergence to differentiate among C. sinensis, trifoliate and C. fortunellae.
With respect of Fortunella it was similar to Citrus in morphology (floral, and fruit) but they both differed in quantitative characters significantly. Pang et al. (2007) possibly explained that Fortunella were of hybrid origin and Citrus might as its putative paternal parent.
Our results clearly showed that close relationship between Fortunella and mandarin and C. reticulata. This results in line with the results of, Kyndt et al. (2010) who supported that Fortunella spp. are clustered within Citrus, close to the C. reticulate.
This study utilized extensive data from SRAP sequence which is more similar to the previous research than the SSR sequence data, thus maybe due to that SRAP targets coding sequences in the genome and the results in a moderate number of co-dominant markers (Agarwal et al., 2008). Consequently, SRAP markers could be more advantageous over SSR markers due to occasional loss of amplification sites of SSR primers in distant Citrus relatives and its relative simplicity (Uzun et al., 2009). Hence, this research provided an opportunity to carefully assess the relative utility of SRAP and SSR divergence.
In conclusion, the present study highlights the usage of SSR and SRAP sequence divergence across DNA level in Citrus and its relatives. To our knowledge, no such studies have been reported yet about comparison of efficiency and ability of SSR and SRAP sequence divergence in Citrus. These results were the new information for future study on citrus breeding programs such as germplasm characterization, screening of zygotic and nucellar seedlings and developing sequence divergence in Citrus and its relatives.
ACKNOWLEDGMENTS
This study was financially supported by the National Natural Science Foundation of China (No. 30921002), the National 863 project, the open fund of the National Key Laboratory of Crop Genetic Improvement and the Biodiversity International (Rome, Italy). The authors thank Dr. Aiman Atta for reviewing the manuscript. Also the authors thank Dr. Rui Zhang, Mr. Jianyong An and Mr. Lijun Chai for their constructive comments and helps.
REFERENCES
- Amar, M.H., M.K. Biswas, Z. Zhang and W.W. Guo, 2011. Exploitation of SSR, SRAP and CAPS-SNP markers for genetic diversity of Citrus germplasm collection. Sci. Horticult., 128: 220-227.
CrossRef - Apostolidis, A.P., Z. Mamuris and C. Triantaphyllidis, 2001. Phylogenetic relationships among four species of Mullidae (Perciformes) inferred from DNA sequences of mitochondrial cytochrome b and 16S rRNA genes. Biochem. Syst. Ecol., 29: 901-909.
PubMed - Agarwal, M., N. Shrivastava and H. Padh, 2008. Advances in molecular marker techniques and their applications in plant sciences. Plant Cell Rep., 27: 617-631.
CrossRefPubMedDirect Link - Biswas, M.K., Q. Xu and X. Deng, 2010. Utility of RAPD, ISSR, IRAP and REMAP markers for the genetic analysis of Citrus spp. Sci. Horticult., 124: 254-261.
CrossRef - Cheng, Y.J., W.W. Guo, H.L. Yi, X.M. Pang and X. Deng, 2003. An efficient protocol for genomic DNA extraction from Citrus species. Plant Mol. Biol. Rep., 21: 177-178.
Direct Link - Chen, C., P. Zhou, Y.A. Choi, S. Huang and F.G. Jr. Gmitter, 2006. Mining and characterizing microsatellites from citrus ESTs. Theor. Appl. Genet., 112: 1248-1257.
Direct Link - Dong, P., Y.M. Wei, G.Y. Chen, W. Li, J.R. Wang, E. Nevo and Y.L. Zheng, 2010. Sequence-related amplified polymorphism (SRAP) of wild emmer wheat (Triticum dicoccoides) in Israel and its ecological association. Biochem. Systematics Ecol., 38: 1-11.
CrossRef - Froelicher, Y., W. Mouhaya, J.B. Bassene, G. Costantino, M. Kamiri, F. Luro, R. Morillon and P. Ollitrault, 2011. New universal mitochondrial PCR markers reveal new information on maternal citrus phylogeny. Tree Genet. Genomes, 7: 49-61.
CrossRef - Gmitter, F.G. Jr., 1995. Origin, evolution and breeding of the grapefruit. Plant Breed. Rev., 13: 345-363.
Direct Link - Guo, W.Z., D. Fang, W.D. Yu and T.Z. Zhang, 2005. Sequence divergence of microsatellites and phylogeny analysis in tetraploid cotton species and their putative diploid ancestors. J. Integr. Plant Biol., 47: 1418-1430.
CrossRef - Kota, R., R.K. Varshney, T. Thiel, K.J. Dehmer and A. Graner, 2001. Generation and comparison of EST-derived SSRs and SNPs in barley (Hordeum vulgare L.). Hereditas, 135: 145-151.
Direct Link - Kyndt, T., T.N. Dung, P. Goetghebeur, H.T. Toan and G. Gheysen, 2010. Analysis of ITS of the rDNA to infer phylogenetic relationships among Vietnamese Citrus accessions. Genet. Resour. Crop Evol., 57: 183-192.
CrossRef - Li, G. and C.F. Quiros, 2001. Sequence-Related Amplified Polymorphism (SRAP), a new marker system based on a simple PCR reaction: its application to mapping and gene tagging in Brassica. Theoret. Applied Genet., 103: 455-461.
CrossRefDirect Link - Liu, Y.Z. and X.X. Deng, 2007. Citrus breeding and genetics in China. Asian Aust. J. Plant Sci. Biotechnol., 1: 23-28.
Direct Link - Luro, F., G. Costantino, J. Terol, X. Argout and T. Allario et al., 2008. Transferability of the EST-SSRs developed on Nules clementine (Citrus clementina Hort ex Tan) to other Citrus species and their effectiveness for genetic mapping. BMC Genomics, 9: 287-287.
CrossRefDirect Link - Dianxiang, Z. and D.J. Mabberley, 2008. 21 CITRUS Linnaeus, Sp. Pl. 2: 782. 1753. Flora China, 11: 90-96.
Direct Link - Makowsky, R., C.L. Cox, C. Roelke and P.T. Chippindale, 2010. Analyzing the relationship between sequence divergence and nodal support using Bayesian phylogenetic analyses. Mol. Phylogenet. Evol., 57: 485-494.
CrossRef - Nicolosi, E., Z.N. Deng, A., Gentile, S. La Malfa, G. Continella and E. Tribulato, 2000. Citrus phylogeny and genetic origin of important species as investigated by molecular markers. Theor. Applied Genet., 100: 1155-1166.
Direct Link - Palmieri, D.A., V.M. Novelli, M. Bastianel, M. Cristofani-Yaly and G. Astua-Monge et al., 2007. Frequency and distribution of microsatellites from ESTs of citrus. Genet. Mol. Biol., 30: 1009-1018.
CrossRef - Pang, X.M., C.G. Hu and X.X. Deng, 2007. Phylogenetic relationships within Citrus and its related genera as inferred from AFLP markers. Genet. Resour. Crop Evol., 54: 429-436.
CrossRef - Scora, R.W., 1975. On the history and origin of citrus. Bull. Torrey Bot. Club, 102: 369-375.
Direct Link - Barrett, H.C. and A.M. Rhodes, 1976. A numerical taxonomic study of affinity relationships in cultivated citrus and its close relatives. Syst. Bot., 1: 105-136.
Direct Link - Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics, 123: 585-595.
PubMedDirect Link - Tamura, K. and M. Nei, 1993. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol., 10: 512-526.
CrossRefPubMedDirect Link - Tamura, K., M. Nei and S. Kumar, 2004. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. U.S.A., 101: 11030-11035.
CrossRefDirect Link - Tamura, K., J. Dudley, M. Nei and S. Kumar, 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol., 24: 1596-1599.
CrossRefPubMedDirect Link - Terol, J., A. Conesa, J.M. Colmenero, M. Cercos and F. Tadeo et al., 2007. Analysis of 13000 unique Citrus clusters associated with fruit quality, production and salinity tolerance. BMC Genomics, Vol. 8.
CrossRef - Thompson, J.D., T.J. Gibson, F. Plewniak, F. Jeanmougin and D.G. Higgins, 1997. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res., 25: 4876-4882.
CrossRefPubMedDirect Link - Uzun, A., T. Yesiloglu, Y. Aka-Kacar, O. Tuzcu and O. Gulsen, 2009. Genetic diversity and relationships within Citrus and related genera based on sequence related amplified polymorphism markers (SRAPs). Sci. Horticult., 121: 306-312.
CrossRef