
Ghalib- Shalaldeh
National center for Agriculture and Technology Transfer, Jordan
Biotechnology
Year: 2006 | Volume: 5 | Issue: 4 | Page No.: 508-513







ABSTRACT
Two methods were evaluated for the extraction of DNA from barley (Hordeum vulgare L.). A CTAB-based method and a nuclei isolation and extraction method. Results showed that barley DNA extracted by nuclei genomic DNA method was of a high molecular weight (HMW) with a very slight shearing. Whereas, DNA extracted by CTAB method gave a high molecular weight (HMW) with the heaviest DNA shearing. All nuclei DNA extracted samples did not show RNA contamination. However, all CTAB extracted samples, showed RNA contamination. Results obtained from the gel of Pulse Field Gel Electrophoresis (PFGE) and Agarose gel electrophoresis were almost similar except for High-LoTM DNA marker (60-10000 bp) that did not appeared in PFGE due to the long period of the run. Furthermore, the two DNA extraction procedures seemed to be simple and efficient in extracting genomic DNA. But, the nuclei method is more effective in extracting intact and HMW of genomic DNA.
PDF Abstract XML References
How to cite this article
Ghalib- Shalaldeh, 2006. Influence of Two Genomic Dna Extraction Methods on Dna Quantity and Quality Extracted from Barley in Jordan. Biotechnology, 5: 508-513.
DOI: 10.3923/biotech.2006.508.513
URL: https://scialert.net/abstract/?doi=biotech.2006.508.513
DOI: 10.3923/biotech.2006.508.513
URL: https://scialert.net/abstract/?doi=biotech.2006.508.513
INTRODUCTION
Barley is one of the most important field crops in Jordan with annual average production during the last year of 34524.2 metric tons (FAOSTAT, 2005). Rapid progress in cultivar improvement through traditional breeding methods is restricted because the process is costly and labor intensive. Therefore, molecular breeding approaches provide additional tools in the improvement of these field crops. The cloning of important genes requires the extraction of large insert genomic DNA. The isolation of intact, high-molecular-mass genomic DNA is essential for many molecular biology applications including long PCR, endonuclease restriction digestion, Southern blot analysis and genomic library construction (Michiels et al., 2003). Many protocols are available for the extraction of DNA from plant material. A cetyltrimethylammonium bromide (CTAB) protocol has been developed for isolation of genomic DNA. In a more commonly used procedure, polysaccharide co precipitation is avoided by adding a selective precipitant of nucleic acids, i.e., cetyltrimethylammonium bromide (CTAB) to keep polysaccharides in solution (Doyle and Doyle, 1987, 1990). Several successful DNA extractions methods for plant species containing polyphenolic compounds and polysaccharides have been developed (Tel-Zur et al., 1999, Peterson et al., 1997, Aljanabi et al., 1999; Burr et al., 2001) and its modifications (Huang et al., 2000). Some publications demonstrated the use of hydrolytic enzymes (Rether et al., 1993) or ion-exchange resins (Guillemaut and 1992 Marechal and Guillemaut, 1995) to remove polysaccharides from nucleic acid solutions. CTAB DNA purification methods extract high quantities of pure DNA from a variety of different plant tissues, e.g., cotton, blackcurrant, ferns, fruit trees and conifers (Woodhead et al., 1998; Dempster et al., 1999; Kim et al., 1997).
A simple and rapid method based on CTAB extraction for isolating high-quality intact DNA was established by modifying existing methods. With this technique, the absorbance ratio (A260/A280) of DNAs obtained from fresh and/or dried roots of medicinal plant (ginseng) tissues enriching with polyphenols and polysaccharides was 1.8 approximately. The results demonstrated that the modified technique was efficient and reliable in isolating high-quality and high-molecular-weight DNAs (Luo et al., 2001). Improved CTAB method, low pH medium with high salt method (LPHS) and urea method were used for DNA extraction. These methods produced high quantity and quality of DNA (Zhang et al., 2004). A quick, simple and reliable CTAB DNA extraction method (method is a modification of Doyle and Doyle (1990). Modified by the use of NaCl to remove polysaccharides and PVP to eliminate polyphenols during DNA purification. Results showed that DNA yield from this procedure is high (up to 1 mg g-1 of leaf tissue) (Muhammad et al., 1994).
Rogers and Bendich (1988) obtained low yields of grapevine DNA with a CTAB-based extraction procedure. Collins and Symons (1992) developed a method that could successfully isolate nuclear DNA from fresh leaves of grapevine, but it was very arduous and the yield was also low. Four DNA extraction protocols (two protocols represented classical procedures and two were a combination of classical CTAB lysis followed by anion exchange chromatography) were evaluated for ability to extract DNA from several species: oak, elm, pine, fir, poplar and maize (fresh materials). Test results indicated superiority of the combination treatment which enabled DNA extraction from all seven species (Csaikli et al., 1998).
Unlike conventional DNA preparation like the CTAB method, the mega base DNA must be protected from physical shearing during preparation. One of the popularly used approaches for doing so is to isolate protoplasts (plants), cells (animals), or nuclei (plants and animals and then embed them in low-melting-point (LMP) agarose in the form of plugs or micro beads (Zhang, 2000). The agarose acts as a solid yet porous matrix, which allows the diffusion of various reagents for DNA purification and subsequent manipulations while preventing the DNA from being sheared (Schwartz and Cantor, 1984). Although the protoplast method yields mega base-size DNA of high quality, the process is costly and labor intensive (Ganal and Tanksley, 1989). However, recently developed nuclei method works well for several divergent plant taxa for its simplicity and cost-effectiveness to isolate high molecular weight DNA (Zhang et al., 1995; Zhang, 2000). The predominant problems involved in trying to isolate plant nuclear DNA are ones that animal researchers do not typically encounter. For example, (a) plant cell walls must be physically broken or enzymatically digested without damaging nuclei, (b) chloroplasts must be separated from nuclei and/or destroyed, (c) volatile secondary compounds such as polyphenols must be prevented from interacting with the nuclear DNA and (d) carbohydrate matrices that often form after tissue homogenization must be prevented from trapping nuclei (Peterson et al., 1997). Large amounts of polyphenolics in cotton leaves make it difficult to obtain high quality genomic DNA during extraction. A procedure to isolate nuclear DNA was developed. It consists of rapid isolation of stable nuclei, which hinders covalent interactions with phenolics, followed by DNA extraction. The yield and quality of the resulting DNA is satisfactory and the protocol can be scaled up or down according to sample size (Chaudhry et al., 1999). Hein et al. (2005) developed a novel nuclei extraction method that allows for the extraction of high molecular weight DNA from leaves of woody perennial soft-fruit species that contain high levels of carbohydrates and polyphenolics. It utilized a modified buffer system including 4% (w/v) (PVP)-10 and a combination of nylon filters and Percoll gradients to purify nuclei extracts prior to embedding in agarose plugs. Extracted DNA was of high quantity and quality suitable for bacterial (BAC) library construction. Wang et al. (1996) adapted the procedure of isolating nuclear DNA from the silica-gel dried leaves with the following improvements (as high as 6% PVP and 2% b-mercaptoethanol were employed to avoid oxidation of polyphenols and 2.5 M NaCl effectively removed polysaccharide rides). They were able to isolate nuclear DNA from the silica-gel dried leaves. Nuclei were separated by low-speed centrifugation (4000 g) from cytoplasm containing secondary compounds. Most genomic DNA extraction protocols require the use of liquid nitrogen and/or freeze-drying (lypholization). A very simple, fast, universally applicable method to extract high quality mega base genomic DNA was applied to extract complex genomic DNA from different (fresh tissues) from wheat, barley, potato, beans, pear and almond leaves, fungi, insects and shrimps. The amount of tissue required by this method is 50-100 mg. The quantity and the quality of the DNA extracted by this method were very high (Salah and Iciar., 1997). The objectives of this study are:
| • | To evaluate the CTAB and the nuclei isolation and extraction methods in extracting high molecular Weigh (HMW) genomic DNA. |
| • | To compare the two methods. |
| • | To detect differences and/or similarities between the results obtained by Pulse Field Gel Electrophoresis (PFGE) and the Agarose Gel Electrophoresis. |
MATERIALS AND METHODS
Plant materials: The certified barley (Hordeum vulgare L) cultivar ACSAD 176 was used in this study. Seeds were germinated and grown in polystyrene tray under greenhouse conditions. One month old, fresh and healthy leaves were used in nuclei isolation and high molecular weight DNA preparation. The upper third of leaves blade was cut and used.
Reagents: SEB (Sucrose-based Extraction Buffer): 10% v/v TKE (0.1M Tris, 1.0 M KCl, 0.1M EDTA pH 9.1), 500 mM sucrose, 4 mM spermidine, 0.1% w/v ascorbic acid, 2.0% w/v PVP (polyvinylpyrrolidone. MW 40, 000) and 0.13% w/v sodium diethyldithiocarbamate.
SEB+BME: 0.2% v/v â-mercaptoethanol (BME) in SEB. β-mercaptoethanol was added just before use.
Lysis Buffer: 1% w/v sodium lauryl sarcosine, 0.1 mg/ml proteinase K, 0.1% w/v ascorbic acid, 6 mM EGTA (Ethylene glycol tetra acetic acid ), 200 mM L-lysine and 0.13% w/v sodium diethyldithiocarbamate in 500 mM EDTA ,pH 9.1. Proteinase K was added just before use.
Nuclei isolation: Nuclei were prepared from barley plants according to Zhang et al. (1995), with some modifications. About 20 g of seedlings was ground into a fine 20 g of fresh leaves tissue were ground into powder in liquid nitrogen with a mortar and pestle. The powder was immediately transferred into 200 mL ice-cold SEB+BME in 250 mL beaker and incubated on ice for 12 min with gently swirling for 20 sec every 2 min. The homogenate was filtered two times through 4 layers of cheesecloth into new ice-cold 250 mL flask. Ten milliliter Triton X-100 (final concentration 5% v/v) was added to the homogenate and incubated on ice for 10 min with gently swirling for 20 sec every 2 min. The mixture was transferred into three 85 mL tubes and centrifuged with fixed-angle rotor at 650 x g for 15 min at 4°C. The supernatant was gently decanted and 10 mL of SEB+BME was added. The pellet was resuspended with assistance of small paint brush. The nuclei suspensions were consolidated into one 85 mL tube then, SEB+BME was added to complete its volume. The nuclei were pelleted by centrifuge at 650 x g for 15 min at 4°C. The supernatant was gently decanted and the pellet was resuspended in 20 mL of SEB. The nuclei were pelleted by centrifuge as described above. All but 1-2 mL of the supernatant was removed with a large pipette. The pellet was resuspended in the residual SEB.
Agarose-embedded nuclei preparation: The nuclei were prewarmed to 45°C in incubator for 10 min before being embedded in agarose. To embed nuclei in agarose plugs, the nuclei were mixed with an equal volume of 1.5% low melting point agarose (BioRad) in SEB using large-bore tip pipette. The nuclei/agarose mixture was aliquot into pre-chilled plug mold (BioRad) on ice with standard yellow tip with its tip cutoff. The agarose was allowed to completely solidify at 4°C for 30 min while still on ice. The solidified plugs were transferred to 50 mL of lysis buffer and incubated at 50°C for 48 h with the lysis buffer replaced after 24 h. Then, the plugs were incubated overnight in 50 ml of fresh Lysis buffer at 4°C. Two plugs from each species were retained in lysis buffer and the resist were equilibrate for four hours with 50 mL of 70% ethanol at room temperature before being stored at -20°C in 70% ethanol.
Preparation of (1% TBE-Agarose gel): Weight 1.5 g of Pulse field certified agarose- BioRad, put them in flask, add 150 ml of 0.5X TBE buffer. Dissolve the agarose using microwave for 1 min to melt the agarose untila clear transparent solution is obtained. Poured the melted solution into a gel casting apparatus (14x13 cm) with 15-tooth gel comb inserted. Allowed the gel to solidify. Stain the gel by ethidium bromide. In suitable container, add 5μL of ethidium bromide to 50 mL 0.5 X TBE buffer. Cover the container with lightproof cover. Allow to stain on a rocker shaker for 30 min. Distain by distilled water for 30 min (10-20 is enough) on rocker shaker. Reset the running setting: Block = 1, Volts = 6 v cm-1, initial switch time = 5 sec, final switch time = 15 sec, running time = 20 h. Transfer to photo documentary unit.
5- DNA extraction by CTAB (Cetyl trimethyl ammonium bromide) method following (Doyle and Doyle,1990):
| • | Reagents: CTAB 2 % v/v, Tris-HCl, pH 8100 mM, EDTA, pH 8 25 mM, NaCl 1.5 M, PVP 1.0 % w/v, β-mercaptoethanol 0.2 % w/v. |
| • | Preparation of CTAB extraction buffer (In 10 mL falcon tube put about 4 mL sterilized H2O, add the above constituents, dissolve and mix by vortex. Bring the volume to 10 mL by sterilized H2O). |
| • | RNase preparation (Put the RNase tube in boiling water for 5 min. then on ice until using). |
DNA extraction by CTAB (Cetyl Trimethyl Ammonium Bromide) method from barley plants is implemented according to Doyle and Doyle (1987), with some modifications. About 4 g of leaves was ground into powder in liquid nitrogen with a mortar and pestle. The powder was immediately transferred into 2 mL tube, add 1.6 mL CTAB extraction buffer. Incubate for 60 min. at 60°C in hybridization oven. It is supplied by rotors that continuously mix the sample, other wise, mix by hand every 2 min. Centrifuge at 4000 rpm for 5 min at 4°C. Take the supernatant (H2O phase) from barley into 2 new 2 mL tubes. Add 0.8 mL of chloroform: octanol (24:1) under fume hood. Shake tubes by vortex for 10 sec. Centrifuge at 4000 rpm for 5 min at 4°C. Take the supernatant (H2O phase) into new 2 mL tube. Add boiled RNase (from 10 mg mL-1 stock) to a final concentration of 20 μg mL. Incubate at RT for 30 min. Re-extract with 0.8 mL of chloroform: octanol (24:1) under fume hood. Shake tubes by vortex for10 sec. Centrifuge at 4000 rpm for 5 min at 4°C. Take the supernatant (H2O phase) into new 2 mL tube. It was light green in color. Add 0.6 volume ice cold isopropanole. Tubes were incubating at -20°C for 10 min. Centrifuge for 10 min at 4°C at 14000 rpm. Discard supernatant. Wash the pellet with 50 μL of 70% ethanol then centrifuge for 10 min at 4°C at 14000 rpm. Discard ethanol and air dry in the Laminar-Flow. Resuspend the pellet in 50 μL of sterile distilled water and Store at 20°C until needed. The tubes were labeled B1, B2.
Agarose mini-gel (0.7%) preparation in TAE buffer (Use 15 teeth comb): Weight 0.5 g of agarose, put them in flask and add 50 mL of 1 X Tris-acetate buffer (TAE buffer). Dissolve the agarose using microwave for 1 min to melt the agarose until a clear transparent solution is obtained. Add ethidium bromide 5 μL (it is added when the temperature become around 40°C). The melted is poured solution into a mould with 15 teeth comb inserted. When gel solidifies, remove the comb in the laminar hood. Take barley plugs out the lysis buffer. Insert the plugs in pre designated gel wells. Cover the plugs with milted LMP agarose. Prevent air bubbles. Put the gel in the electrophoresis apparatus filled (to the required height) with the previous buffer (TAE running buffer). Run at 120 v for 30 min. Transfer to photo documentary unit
RESULTS AND DISCUSSION
Agarose Gel Electrophoresis: DNA yield was determined by the tow methods described earlier (Fig. 1). Marker banding pattern was good. Barley plug (DNA extracted by nuclei method) showed large DNA quantity at the gel well and slightly extended smear. This indicates presence of high molecular weight DNA. Whereas, barley extracted by CTAB first sample (B1) gave heavy smear from the gel well to 10 kbp and less shiny smear from 10 to 1.4 kbp indicating DNA shearing, but still the yield of DNA is high this came in agreement with the results of (Woodhead et al., 1998; Dempster et al., 1999; Kim et al., 1997; Luo et al., 2001; Zhang et al., 2004; Muhammad et al., 1994; Csaikli et al., 1998). But, barley extracted by CTAB the second sample (B2) gave almost one band slightly above 10 kbp. All CTAB extracted samples, showed RNA contamination. CTAB extracted barley sample (B1), showed higher DNA quantity due to less DNA shearing when compared to sample (B2), this disagreed with results obtained by (Rogers and Bendich, 1988; Collins and Symons, 1992) who indicated low yields of DNA with a CTAB-based extraction procedure. Nuclei extraction method for barley (plugs) seem to be more efficient in extracting high molecular weight DNA due to the very rare shearing effect of this method as compared to CTAB method. These results agreed with results obtained by (Schwartz and Cantor, 1984; Zhang, 2000; Zhang et al., 1995, 2004, 2000; Chaudhry et al., 1999; Salah and Iciar, 1997; Hein et al., 2005) who reported higher molecular weight DNA extracted by nuclei method with less shearing effects.
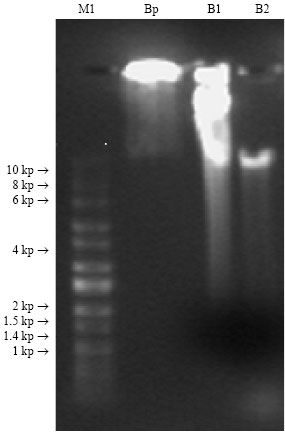 | |
| Fig. 1: | Agarose gel electrophoresis of genomic DNA for: Nuclei isolation DNA (plugs) extraction and CTAB extraction methods from barley: M1: High-LoTMDNA marker (60-10000 bp), Bp: barley plug, B2 and B1: barley extracted by CTAB. Gel run at 120 voltages for 30 min |
Pulse field gel electrophoresis: DNA yield was determined by the tow methods described above (Fig. 2). Low rang marker gave bands similar to the brochure. High-loTM marker gave no bands because it escaped outside the gel due to the long period of the run in the PFGE apparatus. Wheat Wheat extracted by CTAB method showed no bands appearance indicating that the DNA is highly sheared. Barley extracted by nuclei method (plug) showed high DNA at the gel well and smear between the gels well and 200 kbp. This indicates a good DNA quantity or the DNA is not move freely. Barley extracted by CTAB the second sample (B2) showed smear from the gel well to the end but very shiny from≈50 kbp to lower size. However, barley extracted by CTAB first sample (B1) exhibited a very shiny and thick long smear from around 50 kbp to lower weight size.
 | |
| Fig. 2: | Pulse Field Gel Electrophoresis of genomic DNA PFGE: Nuclei isolation DNA (plugs) extraction and CTAB extraction methods from: M1: High-Low DNA marker (60-10000 bp), M2: Low Range Marker plug Bp: barley plug, B1 and B2: barley extracted by CTAB. PFGE running settings: Block = 1, Volts = 6 v cm-1, initial switch time = 5 sec, final switch time = 15 sec, running time = 13 h |
In general, the DNA extraction by the nuclei method (plug) revealed a large quantity of DNA that did not leave the gel well as compared to the CTAB extraction method. These results agreed with results obtained by (Schwartz and Cantor, 1984; Zhang et al., 1995; Zhang, 2000; Chaudhry et al., 1999; Salah Iciar, 1997; Hein et al., 2005) who reported higher molecular weight DNA extracted by nuclei method with less shearing effects and with (Rogers and Bendich, 1988; Collins and Symons, 1992) for the CTAB method who indicated low yields of DNA with a CTAB-based extraction procedure. Generally speaking results obtained from the gel of Pulse Field Gel Electrophoresis (PFGE) and Agarose gel electrophoresis are almost similar except for High-Low DNA marker (60-10000 bp) that did not appeared in PFGE due to the long period of the run.
CONCLUSIONS
These DNA extraction procedures promises simplicity, speed and efficiency, both in terms of time and the amount of plant sample required. In addition, these methods do not require expensive facilities for plant genomic DNA extraction.
There are many advantages in using nuclei genomic DNA extraction method to obtain HMW DNA quantity and quality in barley over the CTAB method. From the present study the following are concluded:
| • | DNA run on both gels (Pulse Field Gel Electrophoresis (PFGE) and Agarose gel electrophoresis) were almost similar, except for High-LoTM DNA marker (60-10000 bp) that did not appeared in PFGE due to the long period of the run. |
| • | Barley DNA extracted by both nuclei genomic DNA and CTAB methods showed HMW DNA quantity with a very rare shearing, however, the DNA extracted by CTAB method exhibited a heavy shearing. |
| • | All nuclei extracted genomic DNA did not show RNA contamination. Whereas, all CTAB extracted genomic DNA showed RNA contamination. |
ACKNOWLEDGMENTS
Special thanks are due to University of Jordan for the Core Biotechnology Lab facilities and due to NCARTT for partially support in providing chemicals. The author would like to express his gratitude to Dr. Hussein Migdadi for his kindly support in facilitating the purchasing and movement of chemicals needed to the project. Deep appreciation to Dr. Monther Saddar show for his supervision and for the colleagues, Faddel Ismail and Nasser Dweick for their lab work.
REFERENCES
- Burr, K., R. Harper and A. Linacre, 2001. One-step isolation of plant DNA suitable for PCR amplification. Plant Mol. Biol. Repoter, 19: 367-371.
Direct Link - Chaudhry, B., Y. Afshan, T. Husnain and S. Riazuddin, 1999. . Mini-scale genomic dna extraction from cotton. Plant Mol. Biol. Reporter, 17: 1-7.
Direct Link - Collins, G.G. and R.H. Symons, 1992. Extraction of nuclear DNA from grape vine leaves by a modified procedure. Plant Mol. Biol. Reporter, 10: 233-235.
CrossRef - Csaikl, U.M., H. Bastian, R. Brettschneider, S. Gauch and A. Meir et al., 1998. Comparative analysis of different DNA extraction protocols: A fast, universal maxi-preparation of high quality plant DNA for genetic evaluation and phylogenetic studies. Plant Mol. Biol. Rep., 16: 69-86.
CrossRefDirect Link - Dempster, E.L., K.V. Pryor, D. Francis, J.E. Young and H.J. Rogers, 1999. Rapid DNA extraction from ferns for PCR-based analyses. Biotechniques, 27: 66-68.
Direct Link - Doyle, J.J. and J.L. Doyle, 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull., 19: 11-15.
Direct Link - Ganal, M.W. and S.D. Tanksley, 1989. Analysis of tomato DNA by pulsed-field gel electrophoresis. Plant Mol. Biol. Reporter, 7: 17-28.
CrossRef - Hein, I.S. Williamson, J. Russell and W. Powell, 2005. Isolation of high molecular weight DNA suitable for bac library construction from woody perennial soft-fruit species. Biotechniques, 38: 69-71.
Direct Link - Huang, J., X. Ge and M. Sun, 2000. Modified ctab protocol using a silica matrix for isolation of plant genomic DNA. Biotechniques, 28: 432-434.
Direct Link - Kim, C.S., C.H. Lee, J.S. Shin, Y.S. Chung and N.I. Hyung, 1997. A simple and rapid method of isolation of high quality genomic DNA from fruit trees and conifers using PVP. Nucleic Acids Res., 25: 1085-1086.
CrossRefDirect Link - Luo, Z.Y., G. Zhou, X.H. Chen, Q.H. Lu and W.X. Hu, 2001. [Isolation of high-quality genomic DNA from plants]. Bull. Hunan Med. Univ., 26: 178-180, (In Chinese).
PubMed - Marechal-Drouard, L. and P. Guillemaut, 1995. A powerful but simple technique to prepare polysaccharide-free DNA quickly and without phenol extraction. Plant Mol. Biol. Repoter, 13: 26-30.
CrossRef - Michiels, A., W. Van-den-Ende, M. Tucker, L. Van-Riet and A. Van-Laere, 2003. Extraction of high-quality genomic DNA from latex-containing plants. Anal. Biochem., 315: 85-89.
Direct Link - Lodhi, M.A., G.N. Ye, N.F. Weeden and B.I. Reisch, 1994. A simple and efficient method for DNA extraction from grapevine cultivars and Vitis species. Plant Mol. Biol. Rep., 12: 6-13.
CrossRefDirect Link - Peterson, D.G., K.S. Boehm and S.M. Stack, 1997. Isolation of milligram quantities of nuclear DNA from tomato (Lycopersicon esculentum), a plant containing high levels of polyphenolic compounds. Plant Mol. Biol., 15: 148-153.
Direct Link - Rether, B., G. Delmas and A. Laouedj, 1993. Isolation of polysaccharide-free DNA from plant. Plant Mol. Biol. Repoter, 11: 333-337.
CrossRef - Tel-Zur, N., S. Abbo, D. Myslabodski and Y. Mizrahi, 1999. Modified CTAB procedure for DNA isolation from epiphytic cacti of the genera Hylocereus and Selenicereus (Cactaceae). Plant Mol. Biol. Rep., 17: 249-254.
CrossRefDirect Link - Woodhead, M., H.V. Davies, R.M. Brennan and M.A. Taylor, 1998. The isolation of genomic DNA from blackcurrant Ribes nigrum L. Mol. Biotechnol., 9: 243-246.
PubMed - Zhang, J.H., M. Zheng and W. Yuan, 2004. Isolation of genomic DNA from Cinnamomum cassia Presl. Zhong Yao Cai, 32: 6-9.
Direct Link - Guillemaut, P. and L. Marechal-Drouard, 1992. Isolation of plant DNA: A fast, inexpensive and reliable method. Plant Mol. Biol. Rep., 10: 60-65.
CrossRefDirect Link